








Раптова серцева смерть обумовлена каналопатіями
Раптова серцева смерть(РСС) - смерть, що розвинулась моментально, або наступила протягом години з моменту виникнення гострих змін в клінічному статусі пацієнта (Douglas Zipes, E.Braunwald. Heart disease. 7 edition.2004). Відповідно до статистики, 80% РСС відбувається вдома, 40% РСС незасвідченні або відбуваються під час сну (Swagemakers V., J. Am. Cardiol. 1997). Тому останнім часом до визначення РСС додано примітку – є свідки які бачили “жертву” напередодні, без видимих ознак хвороби.
Раптова смерть людини, зазвичай, зовні абсолютно здорової, - ситуація крайнього емоційного потрясіння для близьких людей, в той час, як для охорони здоров`я та суспільства в цілому на першому місці стоїть питання, як її попередити, особливо серед відносно молодої та працездатної частини населення. Драматизм даного явища обумовлений не тільки його раптовістю, а перш за все його масовістю. Тільки у США щорічно від серцевих причин раптово помирає 300-400 тис. молодого працездатного населення. Серед більшості країн, включаючи Україну, у більшості таких хворих діагноз встановлюється пізно-після перенесених епізодів раптової зупинки серця, або вже після смерті.
В числі випадків причина раптової смерті залишається нез’ясованою, не дивлячись на детальну патоморфологічну експертизу. Припущення того, що суттєва частина цих випадків є наслідком фатальних серцевих аритмій, обумовлених первинними електричними порушеннями в міокарді, знаходить все більше підтвердження. Прогрес в області молекулярної генетики та клітинної електрофізіології дозволив розкрити механізми, які суттєві для розуміння причин раптової серцевої смерті. Спадкові захворювання, які супроводжуються високим ризиком РСС є важливою проблемою сучасної медицини. У більшості випадків захворювання виявляють в осіб, які не мають ознак органічної патології серцево-судинної системи (primary electric heart disease). Ці захворювання є генетично-детерміновані. Найбільш частими являються синдроми подовженого та вкороченого інтервалу QT, синдром Бругада, аритмогенну дисплазію правого шлуночка (АДПШ). Більшість вказаних захворювань успадковується за аутосомно-домінантним типом, що обумовлює очікувати високу частоту випадків захворювань серед кровних родичів пробанда. Однак виражений внутрішньосімейний поліморфізм захворювання не завжди дозволяє точно вияснити статус членів родини хворого, що призводить до затримок в постановці діагнозу, початку лікування, а в ряді випадків накопичення випадків раптової смерті в родині.
Сучасні підходи до діагностики цих захворювань, оцінці ризику раптової серцевої смерті в таких пацієнтів та вибору тактики лікування в значному ступеню базується на інформації про молекулярно-генетичну природу захворювання. Однак слід визнати, що єдина система генетичного обстеження та консультування таких хворих в Україні абсолютно відсутня. В теперішній час відмічається значна гіподіагностика спадкових захворювань, пов`язаних із високим ризиком раптової серцевої смерті. Це можна пояснити як недостатньою інформованістю широкого кола лікарів різних спеціальностей, бо більшість вказаних синдромів були описані в останні 2-3 десятиліття,так і відсутністю розвинутої програми як кардіологічного так і генетичного скринінгу цих захворювань.
Найбільш вивчене спадкове захворювання, яке супроводжується злоякісними шлуночковими порушеннями ритму серця, - синдром подовженого інтервалу QT ( Long QT syndrome - LQTS), зустрічається з частотою 1:3000-1:5000. Труднощі діагностики синдрому обумовлені тим, що виявити захворювання при відсутності клінічних проявів та очевидного сімейного анамнезу можна тільки на основі ЕКГ обстеження. А у випадках так званої, скритої форми, діагностика можлива тільки за допомогою молекулярно-генетичного аналізу членів родини з вже діагностованим в родині LQT. В зв’язку з цими труднощами часті випадки пізньої діагностики, при яких захворювання виявляється тільки після повторних, багатократних синкопальних станів, кожне з яких може закінчитись раптовою смертю хворого. Пацієнти з синдромом подовженого інтервалу QT можуть тривало спостерігатись у неврологів з діагнозом епілепсія. До сьогодні не є рідкими ситуації виявлення сімейного варіанту LQT тільки після раптової серцевої смерті одного з членів родини під час першого в житті приступу втрати свідомості. В 1993р. виявлена перша мутація в гені, який відповідає за калієвий канал. На сьогодні відомо, що один клінічний фенотип може виникати внаслідок різних генетичних причин, і навпаки, мутація в одному гені може привести до формування клінічно різних захворювань. Синдром подовженого інтервалу QT об`єднує групу патологій, причина яких в генетичних дефектах-мутаціях в генах, які кодують іонні канали і деякі структурні білки.Традиційно виділяють два фенотипи и більше дев`яти генетичних типів захворювання в залежності від молекулярно-генетичного діагнозу. За останні роки були описані також два фенотипові нових захворювання: синдром Андерсена і синдром подовженого інтервалу QT – синдактилія.
Існує два принципових механізми подовження потенціала дії (ПД),які реалізуються на рівні клітинної мембрани: пригнічення вихідних калієвих токів під час фази реполяризації внаслідок блокади калієвих каналів і порушення інактивації натрієвих каналів, що призводить до персистенції входу іонів натрію на протязі всіх фаз ПД. Обидва механізми поєднані з формуванням ранніх постдеполяризації в системі Гіса-Пуркін`є і міокарді шлуночків. Постдеполяризації індукують тригерну активність, запускаючи поліморфну ШТ типу пірует (TdP). Альтернативним механізмом вважають перевантаження клітини іонами кальцію.
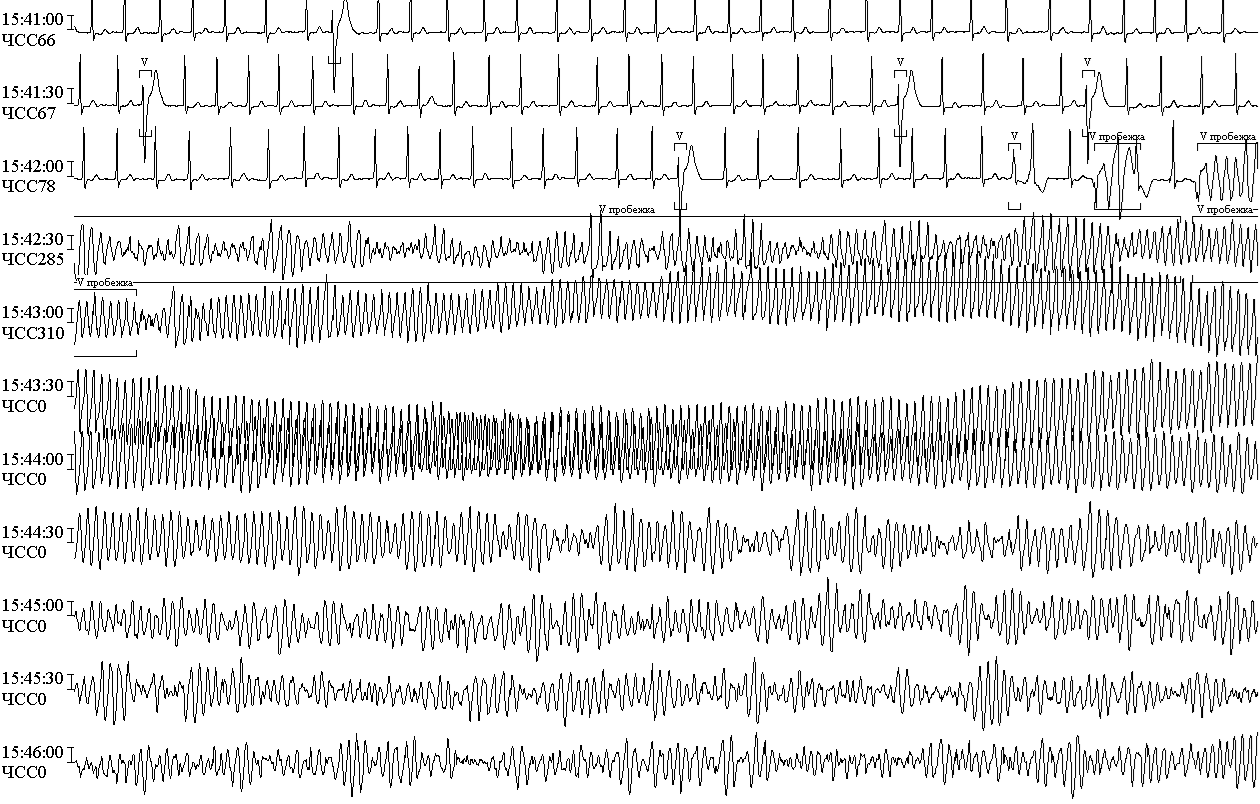
В 1979 році створений міжнародний реєстр первинного (вродженого) синдрому подовженого інтервалу QT.На даний час він об`єднує інформацію більш як про 2000 родин з різними генотипами захворювання. Багатолітнє введення реєстру дозволило отримати важливу інформацію: встановлено, що синдром подовженого інтервалу QT призводить до РСС 3000-4000 дітей та підлітків на рік, причому у осіб чоловічої статі до 15 років вищий ризик розвитку шлуночкової пірует-тахікардії та раптової серцевої смерьі, порівняно із жіночою статтю. Перші ознаки захворювання у жінок виникають пізніше(у 15-40 років), синкопальні стани можуть бути індуковані гормональними змінами, частіше зустрічаються під час менструацій і після пологів. Зрідка захворювання маніфестує у віці 40-50 років, РСС може стати першою клінічною ознакою захворювання. Відомо, що носії гену синдрому подовженого інтервалу QT в популяції зустрічаються з частотою 1:10000. Захворювання нерідко поєднується із бронхіальною астмою.
Перший фенотиповий варіант синдрому подовженого інтервалу QT з аутосомно-рецесивним типом успадкування, синдром Джервела-Ланге-Нільсена (MIM 220400), був описаний у 1957 році. Це захворювання діагностують у дітей із вродженою, двосторонньою глухотою, часто РСС виникає у перші 10 років життя. Описані два генетичних підтипи синдрому в залежності від локалізації мутацій в генах, що кодують білки калієвого каналу, що веде до зниження функції відповідних іонних токів. Ці ж гени експресовані у внутрішньому вусі (продукція ендолімфи), це пояснює поєднання глухоти із подовженням інтервалу QT при даному варіанті синдрому. Другий, фенотиповий варіант синдрому інтервала QT з аутосомно-домінантним типом успадкування, - синдром Уорда-Романо(MIM 192500) описаний незалежно один від одного К.Романо і О Уордом у 60-х роках ХХ століття. На відміну від синдрома Джерела-Ланге-Нільсена при даному захворюванні не буває порушень слуху. Генетичний аналіз виявив суттєву гетерогенність синдрому Романо-Уорда: виявлено шість молекулярно-генетичних типів і більш 400 мутацій у восьми генах, що кодують субодиниці іонних каналів і структурні білки. Функціональні особливості змінених іонних каналів пояснюють відмінності в клінічній картині типів синдрому подовженого інтервалу QT. Перший тип синдрому подовженого інтервалу QT зустрічається частіше інших, характерний мутований ген, який кодує альфа-субодиницю калієвого каналу –відповідального за повільний калієвий тік IKs. Повільний калієвий тік IKs (основний тік реполяризації) активується під дією катехоламінів частотою серцевого ритму. Зниження функції калієвого каналу, відповідального за цей тік, призводить до недостатнього вкорочення інтервалу QT при наростанні частоти. Тому серед хворих з LQTS розвиток шлуночкової пірует-тахікардії майже у 90% випадків виникає в умовах активації симпатоадреналової системи у відповідь на фізичне навантаження та емоційний стрес.
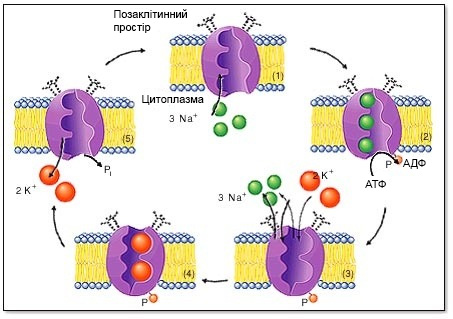
Другий тип синдрому подовженого інтервалу QT (LQTS2), включає 35-45% усіх відомих випадків виникнення синдрому подовженого інтервалу QT. Причиною LQTS2 (MIM 152427) вважають мутацію в гені, який кодує альфа-субодиницю калієвого каналу, відповідального за швидкий калієвий тік IKr. Пароксизми шлуночкової пірует-тахікардії можуть виникати як під час фізичного навантаження чи емоційного стресу, так і в стані спокою. В ролі провокуючих факторів найбільш небезпечні раптові потужні звукові подразнення (грім, крик, сигнал будильника, стукіт у двері).
Третій тип синдрому подовженого інтервалу QT (LQTS3(MIM 603830)) включає 7-15% всіх пацієнтів з синдромом подовженого інтервалу QT. На відміну від перших двох типів, при яких відбувається зниження функції калієвих каналів, при LQTS3 уражується альфа-субодиниця натрієвого каналу, причому дефект веде до посилення функції. Відбувається порушення інактивації натрієвих каналів, виникає постійний вхід іонів натрію, що веде до подовження процесу реполяризації міокарда шлуночків. Фактор, який провокує патологічні зміни при LQTS3 - брадикардія. Пірует-тахікардія при даному типі синдрому подовженого інтервала QT виникає у спокої, переважно під час сну. В цьому випадку збільшення ЧСС має захисну функцію. Пацієнти з LQTS3 добре переносять фізіологічні фізичні навантаження, які ведуть до вкорочення інтервалу QT.
Четвертий тип синдрому LQTS4 - єдиний, при якому відсутня патологія іонних каналів, а мутація відбувається в гені, відповідального за структурний білок анкірин. При LQTS4 подовження інтервалу QT поєднується з вираженою брадикардією, можливо виникнення фібриляції передсердь. При LQTS4 роль фактора провокуючого шлуночкову пірует-тахікардію виконує психоемоційне напруження та фізичне навантаження.
П`ятий та шостий типи синдрому подовженого інтервалу QT (LQTS5 (MIM 176261) і LQTS6(MIM 603796)) - найбільш рідкісні форми захворювання, включають близько 3% усіх LQTS. При LQTS5 мутація виявлена в гені, який кодує бета-субодиницю калієвого каналу, відповідального за повільний калієвий тік IKs. При LQTS6 мутація в гені, який кодує бета-субодиницю калієвого каналу, відповідального за швидкий калієвий тік.
Сьомий тип синдрому подовженого інтервалу QT - LQTS7 частина симптомокомплексу, названого синдромом Андерсена,(MIM 170390), коли LQTS поєднується з періодичним паралічем та дизморфічними змінами. У 1971р. Д. Андерсен описав клінічне спостереження 8-річної дівчинки з гіпертелоризмом, низьким зростом, широкою основою носа, дефектом м`якого та твердого піднебіння та шлуночковими порушеннями ритму серця. Синдром Андерсена включає три основних симптоми: калій-чутливий періодичний параліч, шлуночкові порушення серцевого ритму у вигляді двонапрямленої ШТ, дизморфічні зміни. Розвиток аритмій при LQTS7 провокує гіпокаліємія.
Спадковий синдром подовженого інтервалу QT- синдактилія ( LQTSsyn) - рідкісне захворювання, яке характеризується наступною клінічною картиною: подовження інтервалу QT на ЕКГ, АВ блокада, життєвозагрозливі порушення серцевого ритму, синдактилія, вроджена патологія серця, імунодефіцит, гіпоглікемія, когнітивні аномалії та аутизм. Для даного типу синдрому ідентифікована мутація в гені, відповідального за L-тип кальцієвих каналів-LQTS8.
Гени, що відповідають за розвиток синдрому подовженого інтервалу QT
|
Варіант LQTS |
Локалізація |
Ген |
Білковий продукт |
|
LQT1 |
11p15.5 |
KCNQ1 |
α-субодиниця калієвого каналу (IKs) |
|
LQT2 |
7q35-36 |
KCNH2 |
α-субодиниця калієвого каналу (IKr) |
|
LQT3 |
3p21-24 |
SCN5A |
α-субодиниця натрієвого каналу (INa) |
|
LQT4 |
4q25-27 |
ANK2 |
Анкірин В |
|
LQT5 |
21q22.1-22 |
KCNE1 |
β-субодиниця калієвого каналу (IKs) |
|
LQT 6 |
21q22.1-22 |
KCNE2 |
β-субодиниця калиевого канала (IKr) |
|
LQT 7 |
17q23.1-q24.2 |
KCNJ2 |
α-субодиниця калієвого каналу (IKr) |
|
LQT8 |
12p13.3 |
CACNA1C |
α1-субодиниця кальцієвого каналу L-типу |
|
LQT9 |
3p25 |
CAV3 |
Кавеолін-3 |
|
LQT10 |
11q23 |
SCN4B |
β-субодиниця натрієвого каналу |
|
LQT11 |
7q21-q22 |
AKAP9 |
А-кіназа центросоми і комплексу Гольджі |
|
LQT12 |
20q11.2 |
SNTA1 |
α1-субодиниця синтрофіну |
Ефективність різних методів лікування при різноманітних молекулярно-генетичних варіантах синдрому LQTS.
|
|
LQT1, LQT5 |
LQT2, LQT6 |
LQT3 |
|
Чутливість до симпатичної стимуляції |
+++ |
+ |
- |
|
Обставини, при яких частіше спостерігається пірует-тахікардія |
Фізичне навантаження |
Переляк |
Спокій / сон |
|
Специфічний провокуючий фактор |
Плавання |
Різкий звук, післяродовий період |
- |
|
Обмеження фізичного навантаження |
+++ |
+ |
- |
|
β-блокатори |
+++ |
+ |
- |
|
Препарати калію |
+? |
+++ |
+? |
|
Антиаритмічні препарати IB класу (блокатори натрієвих каналів) |
+ |
++ |
+++ |
|
Блокатори кальцієвих каналів |
++ |
++ |
+? |
|
Блокатори калієвих каналів (нікорандил) |
+ |
+ |
- |
|
Імплантація електрокардіостимулятора (ШВРС) |
+ |
+ |
+++ |
|
Імплантація кардіовертера-дефібрилятора |
++ |
++ |
+++ |
Розроблені П. Дж. Шварц та співав. у 1985р. діагностичні критерії LQTS були відкоректовані у 1993р. До великих критеріїв відносять: подовження інтервалу QT більше за 440 мс; наявність в анамнезі епізодів втрати свідомості(ознака пірует-тахікардії; наявність синдрому подовженого інтервалу QT у членів родини.
Малі критерії: врождена нейросенсорна туговухість; епізоди альтернації зубців Т на ЕКГ; брадикардія у дітей(ЧСС менше вікових норм); патологічна шлуночкова реполяризація у вигляді зубця Т з зазубринкою.
Спадковий синдром подовженого інтервалу QT діагностують при наявності двох великих або одного великого та двох малих критеріїв. Остаточний діагноз виставляють на основі даних молекулярно-генетичного обстеження.
Синдром вкороченого інтервалу QT (ShortQTS)
Синдром вкороченого інтервалу QT був описаний у 2000р. (I.Gussak), спостерігавши трьох членів однієї родини з пароксизмальною формою фібриляції передсердь. Характерні ЕКГ ознаки даного синдрому – тривалість інтервалу QT <300 мс, високий симетричний піскоподібний зубець Т в правих прекордіальних відведеннях. В основі ідіопатичного вкорочення інтервалу QT- спадковий дефект іонних каналів клітин серця. Захворювання успадковується за аутосомно-домінантним типом. Основні клінічні симптоми - синкопальні стани, обумовлені пароксизмами шлуночкової тахікардії, які підвищують ризик розвитку раптової аритмічної смерті. В осіб з даним синдромом були зареєстровані випадки раптової смерті у віці від 3 міс до 71 року. Нерідко виникають пароксизми фібриляції передсердь. Но основі молекулярно-генетичного аналізу виділяють три підтипи цього стану. При всіх підтипах синдрому вкороченого QT відбувається посилення вихідного калієвого току, прискорення процесів реполяризації, вкорочення потенціалу дії і пропорційне зменшення часу рефрактерного періоду міокарда. Вкорочення ефективного рефрактерного періоду несе за собою зменшення довжини хвилі збудження – одного з ключових факторів стійкості феномену re-entry, основи розвитку шлуночкової тахікардії та фібриляції передсердь.
Молекулярно-генетична класифікація синдрому вкороченого інтервалу QT:
|
Захворювання |
Локус |
Ген |
Білок |
Функція білків/вплив мутації |
|
ShortQTS1 |
7q35-q36 |
KCNH2 |
HERG |
IKr-калієвий канал (альфа-субодиниця)/посилення функції |
|
ShortQTS2 |
11p15.5 |
KCNQ1 |
KLVQT1 |
IKs-калієвий канал(альфа-субодиниця)/посилення функції |
|
ShortQTS3 |
17q23 |
KCNJ2 |
Kir 2.1 |
IK1-калієвий канал/посилення функції |
Синдром Бругада (MIM 601144) моногенне полілокусне захворювання, обумовлене мутаціями в гені SCN5А, який кодує альфа-субодиницю натрієвого каналу, клінічно проявляється поліморфною шлуночковою тахікардією та фібриляцією шлуночків в поєднанні із підйомом сегменту ST в правих грудних відведеннях. До розвитку клінічного фенотипу синдрому Бругада призводять мутації в гені SCN5А, що реалізуються за типом “loss of function”. Перше повідомлення про синдром Бругада з`явилось у 1992р., коли брати P.Brugada и J.Brugada описали випадки раптової смерті у молодих чоловіків в країнах Південно-Східної Азії. Сьогодні відомо, що це захворювання зустрічається практично у всіх країнах. Частота синдрому Бругада в Європі та США 1 :10000 населення.
Ідентифіковані мутації в гені SCN5A у хворих із синдромом Бругада:
|
Нуклеотидна заміна |
Зміна білка |
Фрагмент гену |
Ділянка білка |
|
IVS16DS-5A>G |
Мутація сплайсингу |
Інтрон 16 |
DII-DIII |
|
Del2542-2544 |
Del 848I |
Екзон 16 |
DII-DIII |
|
IVS24AS+1G>A |
Мутація сплайсингу |
Інтрон 24 |
DIII-DIV |
На сьогодні недостатньо інформації про те, як саме включати інформацію про генетичну природу захворювання у існуючу стратифікацію ризику раптової серцевої смерті. Однак не викликає сумнівів, що в родинах із вперше виявленим синдромом Бругада має бути проведене молекулярно-генетичне обстеження. У випадку виявлення мутації, ДНК-діагностика показана всім кровним родичам хворого.
Аритмогенна дисплазія правого шлуночка (АДПШ) – генетично детерміноване спадкове захворювання серця, яке характеризується фіброзно-жировим заміщенням міокарда, переважно правого шлуночка, та супроводжується порушеннями ритму серця у вигляді шлуночкової екстрасистолії та правошлуночкової тахікардії з високим ризиком раптової серцевої смерті людей молодого віку, спортсменів.
Аритмогенна дисплазія правого шлуночка була описана у 1977р. Дж. Фонтейном та співав. Виявлені ними хворі, страждали на резистентну до медикаментозної терапії ШТ, але не мали клінічних ознак серцево-судинної патології. Пізніше був встановлений зв`язок аритмогенної дисплазії ПШ з непоясненою раптовою серцевою смертю у молодому віці без ішемічної хвороби серця в анамнезі. Спадкова природа захворювання підтверджується у 30% випадків. За виключенням деяких родин, АДПШ успадковується за аутосомно-домінантним типом. На сьогодні, гени відповідальні за АДПШ не до кінця ідентифіковані, однак виявлено зчеплення даного захворювання з сімома локусами, які картовані на 1-й, 3-й, 10-й, 14-й хромосомах.
Генетичні дефекти, асоційовані з АДПШ:
|
Тип АДПШ |
білок |
Тип успадкування |
Хромосома/ген |
|
АДПШ 1 |
Ріанодіновий рецептор |
Аутосомно-домінантний |
14q24.3/RYR2 |
|
АДПШ 2 |
невідомий |
Аутосомно-домінантний |
1q42 |
|
АДПШ 3 |
невідомий |
Аутосомно-домінантний |
14q11q12 |
|
АДПШ 4 |
невідомий |
Аутосомно-домінантний |
2q32 |
|
АДПШ 5 |
невідомий |
Аутосомно-домінантний |
3p23 |
|
АДПШ 6 |
невідомий |
Аутосомно-домінантний |
10p12p14 |
|
АДПШ 7 |
невідомий |
Аутосомно-домінантний |
10q22 |
|
АДПШ 8 |
Десмоплакін |
Аутосомно-домінантний |
6p28/DSP |
|
Хв. Наксоса |
Плакоглобін |
Аутосомно-домінантний |
17q21/JUP |
|
АДПШ |
Невідомий |
Аутосомно-домінантний |
14q24q |
|
АДПШ |
десмоглобін |
Аутосомно-домінантний |
6p24 |
Основний клінічний симптом АДПШ - шлуночкові аритмії з морфологією ектопічних комплексів по типу блокади лівої ніжки пучка Гіса. На ЕКГ на фоні синусового ритму присутні зміни, характерні порушенням процесів деполяризації та реполяризації міокарда шлуночків, більше в правих грудних відведеннях. Можуть виникати ознаки повної чи неповної блокади правої ніжки пучка Гіса з вторинними змінами кінцевої частини шлуночкового комплексу. Найбільш специфічна ознака патології епсилон-хвиля, яка реєструється в початковій частині сегменту ST. Її вважають інтегральним електрокардіографічним відображенням пізніх потенціалів шлуночків, що вказує на наявність в міокарді зон із різко порушеним проведенням.
Первинні електричні захворювання серця відповідальні за більшу частину РСС в осіб з анатомічно нормальним серцем. Об’єднання цих захворювань в окрему групу обумовлене загальним для них принципом генетично детермінованого високого ризику розвитку шлуночкових аритмій з наступним переходом у фібриляцію шлуночків в осіб без видимих морфологічних змін зі сторони серцево-судинної системи. Для більшості з цих патологічних станів характерні специфічні ЕКГ-аномалії, висока внутрішньосімейна концентрація. Медико-генетичне консультування родин із первинними порушеннями серцевого ритму має проводитись за єдиною схемою. Рекомендації повинні мати конкретний характер, враховувати клінічні прояви захворювання у пробанда, кровних родичів та характер молекулярно-генетичного дефекту. Для накопичення та узагальнення інформації, про клінічні особливості, ризики, підходи до терапії, іхню ефективність, а також для вдосконалення медичної допомоги хворим із первинними порушеннями серцевого ритму необхідне створення єдиного регістру спадковообумовлених порушень серцевого ритму.